Explore Q-Chem Features
Q-Chem is a robust software platform with an extensive set of features. Whether you want to study spin-orbit coupling effects in a single-molecule magnet, run high-throughput calculations on small organic molecules, study an enzyme using QM/MM, or something entirely different, our software package offers a wide range of solutions for a variety of applications. Check out our available features and see how Q-Chem can help you achieve your research goals!

We are pleased to present the sixth major release of the Q-Chem ab inito quantum chemistry software package, Q-Chem 6.1. Our latest release has many improvements and new features, including:
- Force decomposition analysis (Abdulrahman Aldossary, Yuezhi Mao, Marti Gimferrer)
- RI-CC2-EOM-SF, EA, and IP (Garrette Paran, Thomas Jagau, Cansu Utku)
- Improvements to the new libopt3 geometry optimizer, including transition state optimization and new initial model Hessian (Peter McLaughlin)
- Vibrational circular dichroism (VCD) spectroscopy (Yu Zhang, Kuan-Yu Liu, Eric Berquist, Evgeny Epifanovsky)
- Decomposition analysis of vibronic spectra with IQmol (Kuan-Yu Liu, Peter McLaughlin, Andrew Gilbert)
- Implementation of PCM solvent effects for the nuclear-electronic orbital (NEO) method (Mathew Chow, Sharon Hammes-Schiffer)
- Computation of MO overlaps at displaced geometries (John Herbert)
- Spin-orbit NTO analysis for TDDFT and SF-TDDFT (Saikiran Kotaru)
- Addition of POL and NOCV terms to energy decomposition analysis (Hengyuan Shen)
For a complete list of new features, bugfixes, and improvements, please see the Q-Chem 6.1 release log. Interested in trying our new features? Request a free two-month demo here!


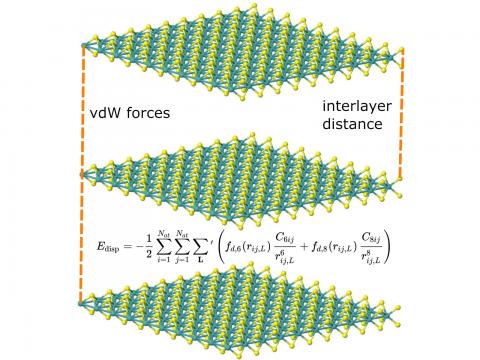
Q-Chem supports LDA, GGA, and meta-GGA functionals, as well as hybrid, range-separated hybrid, and double hybrid versions of both GGAs and meta-GGAs. Single-point energies, geometry optimizations, vibrational frequency calculations, and many other properties can be evaluated for ground states, and for excited states via time-dependent DFT.

Q-Chem offers state-of-the-art tools for treating electron correlation effects, such as Møller-Plesset perturbation theory and coupled-cluster theory. For systems with strong correlation, Q-Chem offers specialty treatments including CASSCF, coupled-cluster valence bond theory, selected CI, RAS-CI, spin-flip, and variational 2-RDM methods.
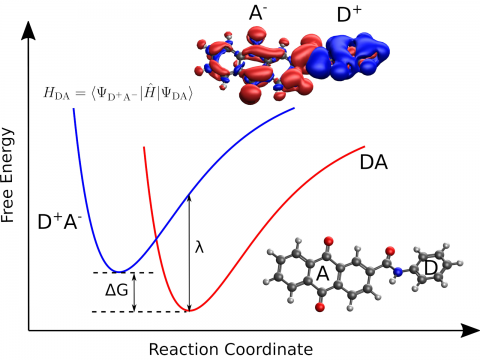
Q-Chem provides a diverse set of methods for studying electronically excited states: CIS, TD-DFT, NOCI, EOM-CC, and ADC. Specialty flavors of these methods cover many types of electronic structure, making it possible to simulate spectroscopic features, charge and energy transfer, and non-adiabatic dynamics. Additionally, our wavefunction analysis module can be used to provide further insight into excited states.
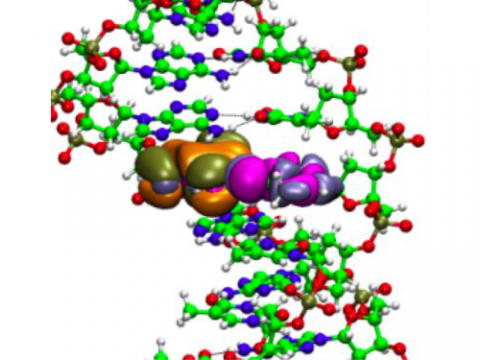
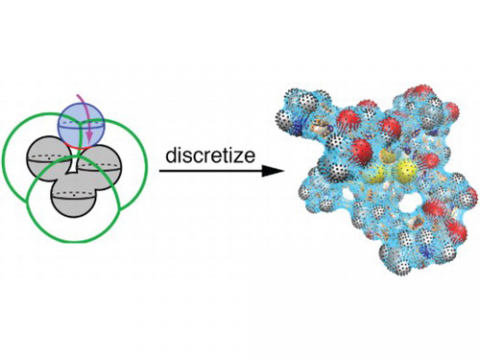
The Q-Chem package offers a variety of solutions for modeling solvated systems, ranging from implicit solvent models, such as SM8, COSMO, and C-PCM, to the effective fragment potential method, which can be used to capture explicit solvent effects. Additionally, Q-Chem includes several different embedding approaches, including QM/MM and density embedding, as well as interfaces to CHARMM and GROMACS.
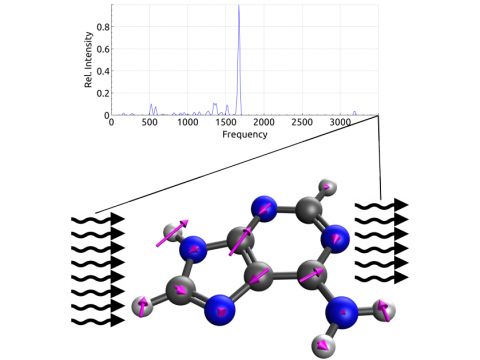
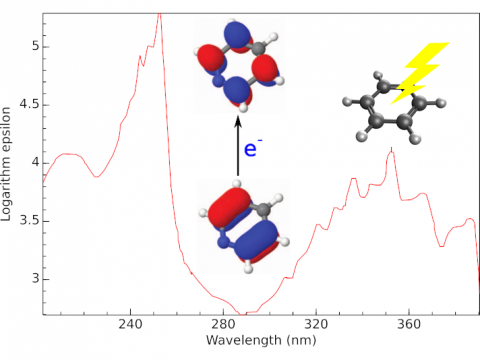
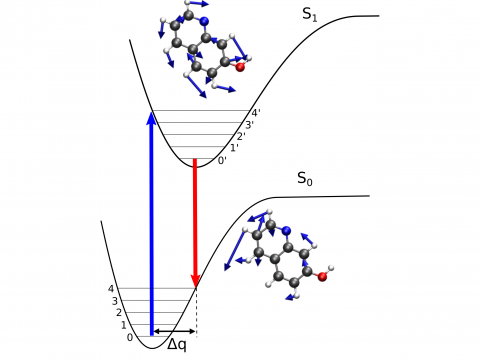
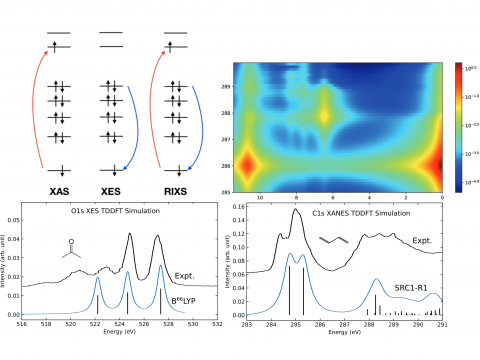
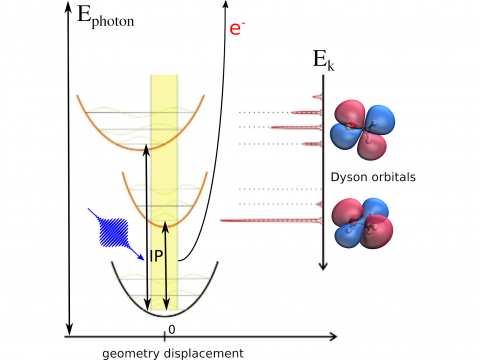
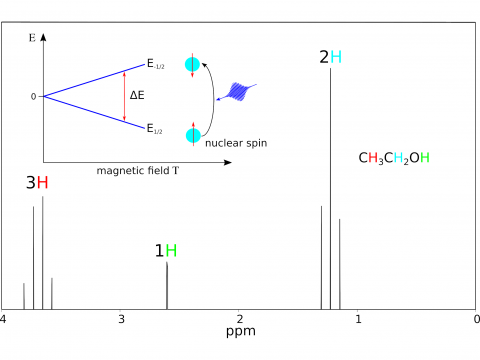
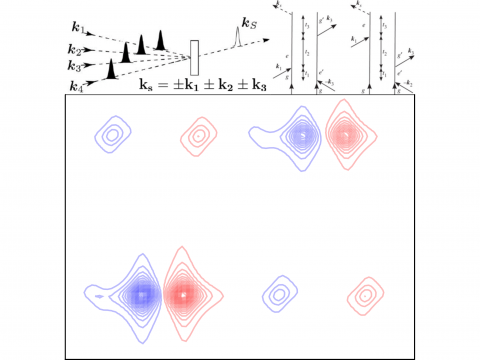
Q-Chem offers a variety of tools for modeling different types of spectra. Our capabilities include IR and Raman spectroscopy, UV-vis spectroscopy, X-ray spectroscopy, photoelectron spectroscopy, NMR spectroscopy, and nonlinear spectroscopy (such as two-photon absorption). Spectroscopic features can be studied using many different levels of theory, ranging from TDDFT to EOM-CC and ADC methods.
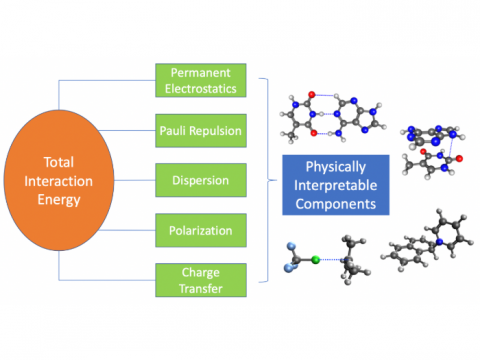
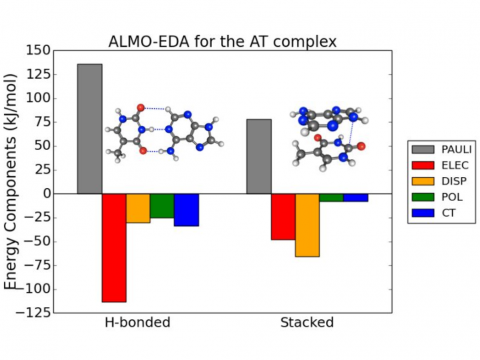
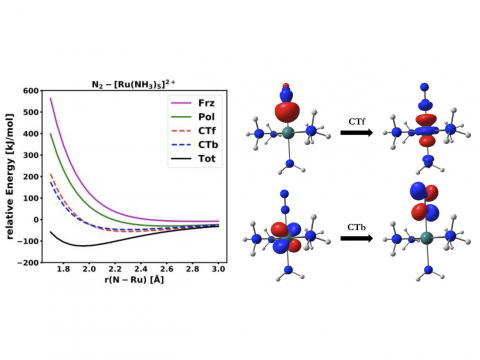
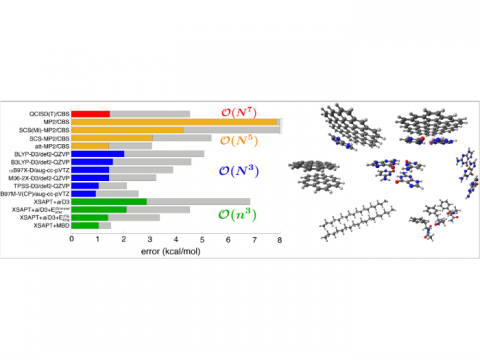
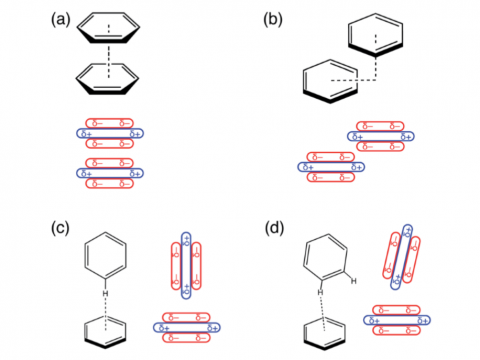
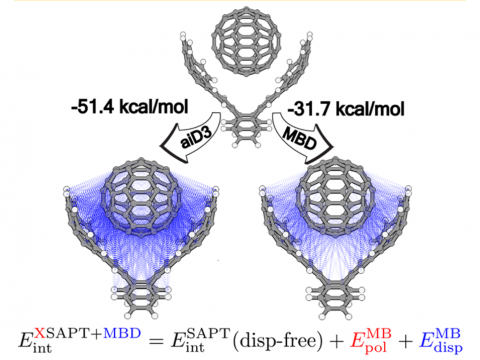
Energy decomposition analysis based on absolutely localized molecular orbitals provides a breakdown of the total interaction energy into meaningful physical terms, providing insights into the nature of intermolecular and bonded interactions. Symmetry-adapted perturbation theory (SAPT) and an extended many-body version thereof (XSAPT) are also available for computing and analyzing intermolecular interactions.

Q-Chem provides methods for geometry optimization, potential energy surface scans, transition state searches, and intrinsic reaction coordinate following, making it ideal for studies of chemical reactivity, thermochemistry, and chemical kinetics.
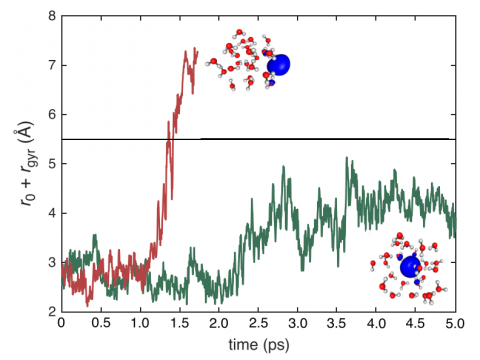
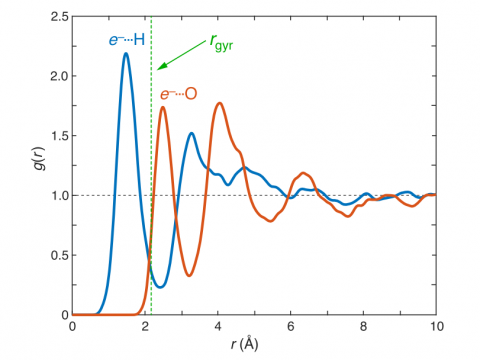
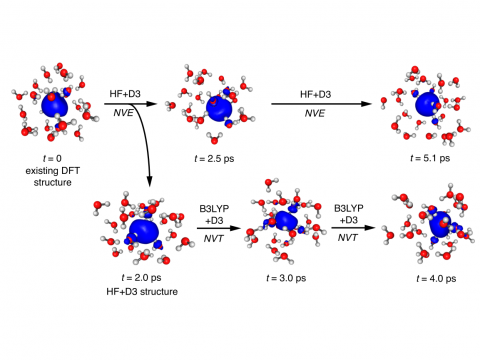
Q-Chem can perform ab initio molecular dynamics (AIMD), including both NVE and NVT thermal samplings, as well as quasi-classical molecular dynamics (QMD). These approaches can be used to produce vibrational spectra and ab initio path integrals. We also include an implementation of Tully's fewest-switches surface hopping (FSSH) approach to effectively handle non-adiabatic systems.