Thermally-Assisted-Occupation Density Functional Theory (TAO-DFT)
The thermally-assisted-occuptation DFT (TAO-DFT) method, now available in Q-Chem, provides accurate descriptions of the ground states of strongly correlated systems. It uses partial orbital occupations, which are defined by a Fermi-Dirac distribution and controlled by a fictitious temperature \(\theta\), to account for multireference character.
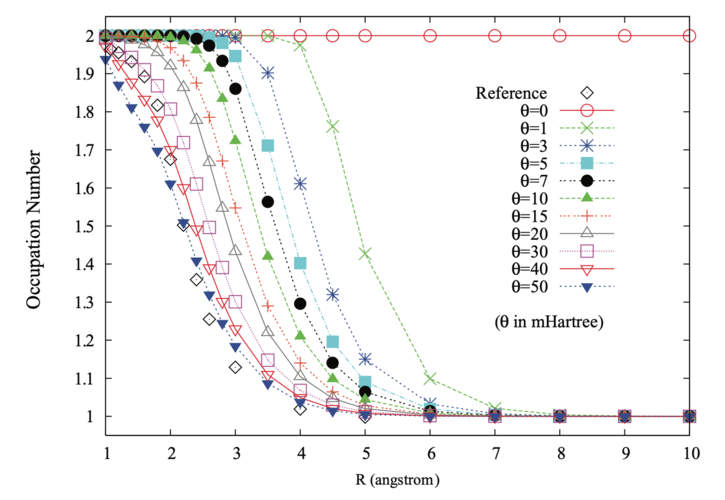 *Occupation numbers of the \(3\sigma_g\) orbital for N\(_2\) compared to MRCI values. *
*Occupation numbers of the \(3\sigma_g\) orbital for N\(_2\) compared to MRCI values. *
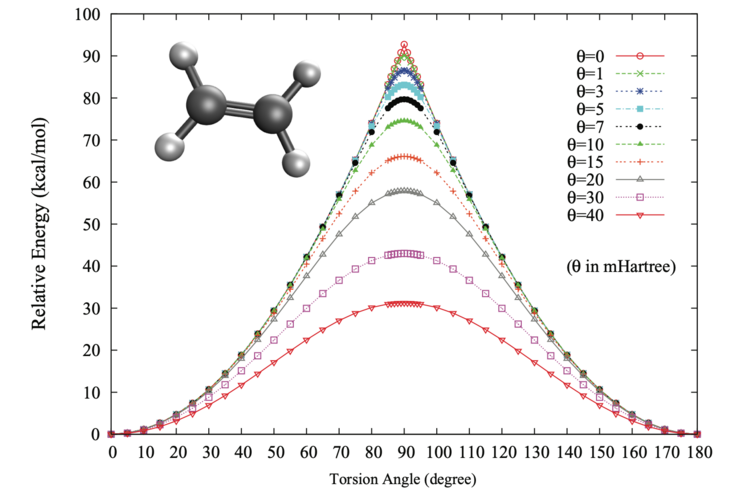 *Potential energy surface for the torision of twisted ethylene. *
*Potential energy surface for the torision of twisted ethylene. *
-
Improved DFT description of the ground states of strongly correlated systems
-
Similar computational cost to traditional KS-DFT for energies and analytical nuclear gradients
-
No pre-determined active space required
-
Can be used with existing XC functionals (e.g. LDA or GGA)
-
Allows simulation of very large, strongly correlated polyradical systems on the nanoscale
-
Can be combined with ab initio molecular dynamics approaches (TAO-AIMD)
Want to try Q-Chem?