Q-Chem Webinar 43
New Analysis Tools for Excited-State Quantum Chemistry: The libwfa Library in Q-Chem

Presented by Felix Plasser on
Felix Plasser was born and raised in Vienna, Austria. In 2012 he earned his Ph.D. at the University of Vienna in the group of Hans Lischka working on the photophysics of interacting nucleobases. In 2013 he moved to Heidelberg to spend two years at Ruprecht-Karls University in the group of Andreas Dreuw to study phosphorescent iridium complexes. He returned to Vienna in 2015 to join the group of Leticia González working on a variety of photoinduced molecular processes. In February 2018, he was appointed to a Lecturer position at the University of Loughborough, UK. Felix Plasser has worked on a wide range of topics in computational photochemistry regarding questions of biological and technological interest. His expertise lies in high-level electronic structure computations combined with detailed wavefunction analysis protocols and dynamics simulations. He has designed and developed the excited-state wavefunction analysis package TheoDORE. Furthermore, he has made significant contributions to the electronic structure packages COLUMBUS, Q-Chem, and MOLCAS as well as the nonadiabatic dynamics packages Newton-X and SHARC. To this date, he has published 67 peer-reviewed articles, which have been cited over 3000 times.
Abstract
Excited electronic states are central to many areas of modern science and computational methods are becoming ever more accurate in their description. However, a new bottleneck is encountered in the analysis of the computations owing not only to the quantity of data produced but also because many of the phenomena studied are difficult to grasp within the standard molecular orbital (MO) picture. To remove this barrier an extended wavefunction analysis framework has been developed over the last years and integrated into Q-Chem [1,2]. We will illustrate how these tools can be used to (i) analyze excited state character in an automated and reproducible way, (ii) bridge between the MO, exciton and valence-bond pictures, and (iii) provide new insight into excited state energetics beyond the MO picture.
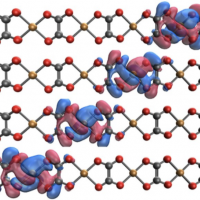
We start by illustrating how excited-state localization and charge transfer in the excited states of organic push-pull systems can be quantified in a completely automated way not requiring any visual inspection of orbitals [3]. Subsequently, we show how the formalism can be extended to quantify charge separation even in centrosymmetric systems and proceed by analyzing electron-hole separation in conjugated polymers. This work sheds new light on fundamental photophysical processes in extended systems while it also gives new insight into the challenges of their description with TDDFT using various functionals [4]. Finally, we outline how the developed methods provide new insight into the connections of the MO and valence-bond pictures using the paradigmatic case of the ionic and covalent states of naphthalene [5] and present a model for explaining the relative energies of the involved states based on their transition densities [2].
References
[1] F. Plasser, M. Wormit, A. Dreuw, J. Chem. Phys. 2014, 141, 024106.
[2] P. Kimber, F. Plasser, PCCP 2020, 22, 6058–6080.
[3] F. Plasser, J. Chem. Ph
Supporting Material

Questions or suggestions?
We hope you enjoyed this webinar! If you have any questions or feedback for the speaker or the Q-Chem team on the topics covered in this webinar, please use this forum thread:https://talk.q-chem.com/t/webinar-43-by-felix-plasser-new-analysis-tools-for-excited-state-quantum-chemistry-the-libwfa-library-in-q-chem/129.