Q-Chem Webinar 60
Orbital Optimized Density Functional Theory for Electronic Excited States

Presented by Diptarka Hait on
Diptarka Hait received his S.B. degree in Chemistry and Physics from MIT in 2016, working in the group of Prof. Troy Van Voorhis and was introduced to the world of OO-DFT. He is currently a PhD candidate in Prof. Martin Head-Gordon's group at the UC Berkeley. He is interested in the development of quantum chemistry methods and their application to problems of interest to the experimental community, with specific emphasis on the use of DFT beyond ground state energetics.
Abstract
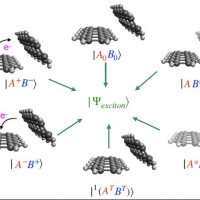
Density Functional Theory-based modeling of electronic excited states is of importance for the investigation of the photophysical/photochemical properties and spectroscopic characterization of large systems. The widely-used linear response Time-Dependent DFT (TDDFT) approach is not effective at modeling many types of excited states, including charge-transfer states, doubly excited states and core-level excitations.
In this webinar, I will discuss the use of state-specific Orbital Optimized (OO) DFT approaches as an alternative to TDDFT for electronic excited states. I will first discuss the motivation and theory behind such approaches, along with some challenges faced by such methods both historically and at present. Subsequently, I will present the Square Gradient Minimization (SGM) algorithm for reliable and efficient excited state orbital optimization, which has been implemented in Q-Chem. In particular, SGM permits use of Restricted Open-shell Kohn-Sham (ROKS) for modeling arbitrary singlet excited states. ROKS/SGM can thus be used to compute core-excitation energies, with the modern SCAN functional yielding ~ 0.3 eV error vs experiment (compared to ~0.1 eV uncertainty in the experimental values themselves). The use of ROKS/SGM in computing Near-Edge X-ray Absorption Fine Structure (NEXAFS) will be discussed. Time permitting, I would also touch upon the recent implementation of the X2C relativistic model in Q-Chem, which (among other things) can be used to accurately compute NEXAFS of elements as heavy as Cr.
Supporting Material
References
- Orbital Optimized Density Functional Theory for Electronic Excited States.
Hait D., Head-Gordon M.
Journal of Physical Chemistry Letters 2021 12 (19), 4517–4529. - Excited state orbital optimization via minimizing the square of the gradient: General approach and application to singly and doubly excited states via density functional theory.
Hait D., Head-Gordon M.
Journal of Chemical Theory and Computation 2020 16 (3), 1699-1710. - Highly Accurate Prediction of Core Spectra of Molecules at Density Functional Theory Cost: Attaining sub eV Error from a Restricted Open-Shell Kohn-Sham Approach.
Hait D., Head-Gordon M.
Journal of Physical Chemistry Letters 2020 11 (3), 775-786. - Accurate prediction of core-level spectra of radicals at density functional theory cost via square gradient minimization and recoupling of mixed configurations.
Hait D., Haugen E.A., Yang Z., Oosterbaan K.J., Leone S.R., Head-Gordon M.
Journal of Chemical Physics 2020 153 (13), 134108.

Questions or suggestions?
We hope you enjoyed this webinar! If you have any questions or feedback for the speaker or the Q-Chem team on the topics covered in this webinar, please use this forum thread:https://talk.q-chem.com/t/webinar-60-by-diptarka-hait-orbital-optimized-density-functional-theory-for-electronic-excited-states/568.