Q-Chem 7: Ab Initio Molecular Modeling
We are pleased to introduce Q-Chem 7! Upgrade today and enjoy improved performance and usability, as well as new tools for studying chemistry and spectroscopy. Deploy large-scale calculations and workflows, get accurate results faster, and extend the scope of your research to exciting new research questions.

Highlighted Features In Q-Chem 7
COACH Functional
COACH is a new range-separated hybrid (RSH) meta-GGA functional that is more accurate and generally transferable than the best existing RSH meta-GGAs, such as ωB97M-V. Read the paper here.
Linear Scaling via Local Correlation
Q-Chem 7 includes a new linear-scaling local correlation algorithm for MP2 and double-hybrid DFT! It is faster and more accurate than DLPNO and uses a single numerical threshold, making it easy to tune accuracy. Read the paper here.
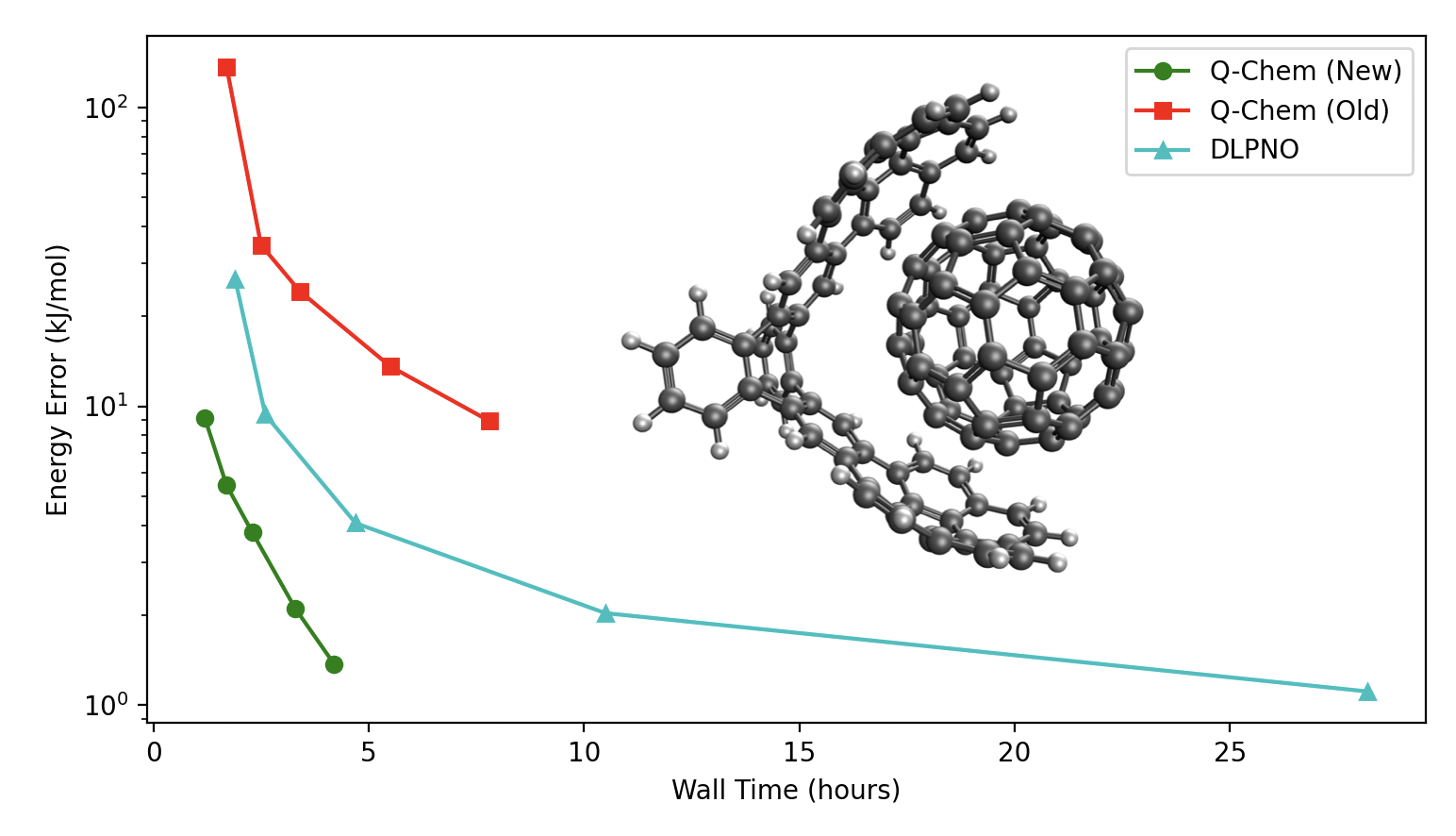
Comparison of interaction energy error and compute time between Q-Chem's regular MP2, the new LMP2 available in Q-Chem 7, and DLPNO-MP2.
More Cutting-Edge DFT Features
- Ongoing performance improvements, including RI speedup and MPI
- “Robust SCF” for improved convergence
- New complex-variable functionals
- Complex absorbing potentials (CAP-DFT)
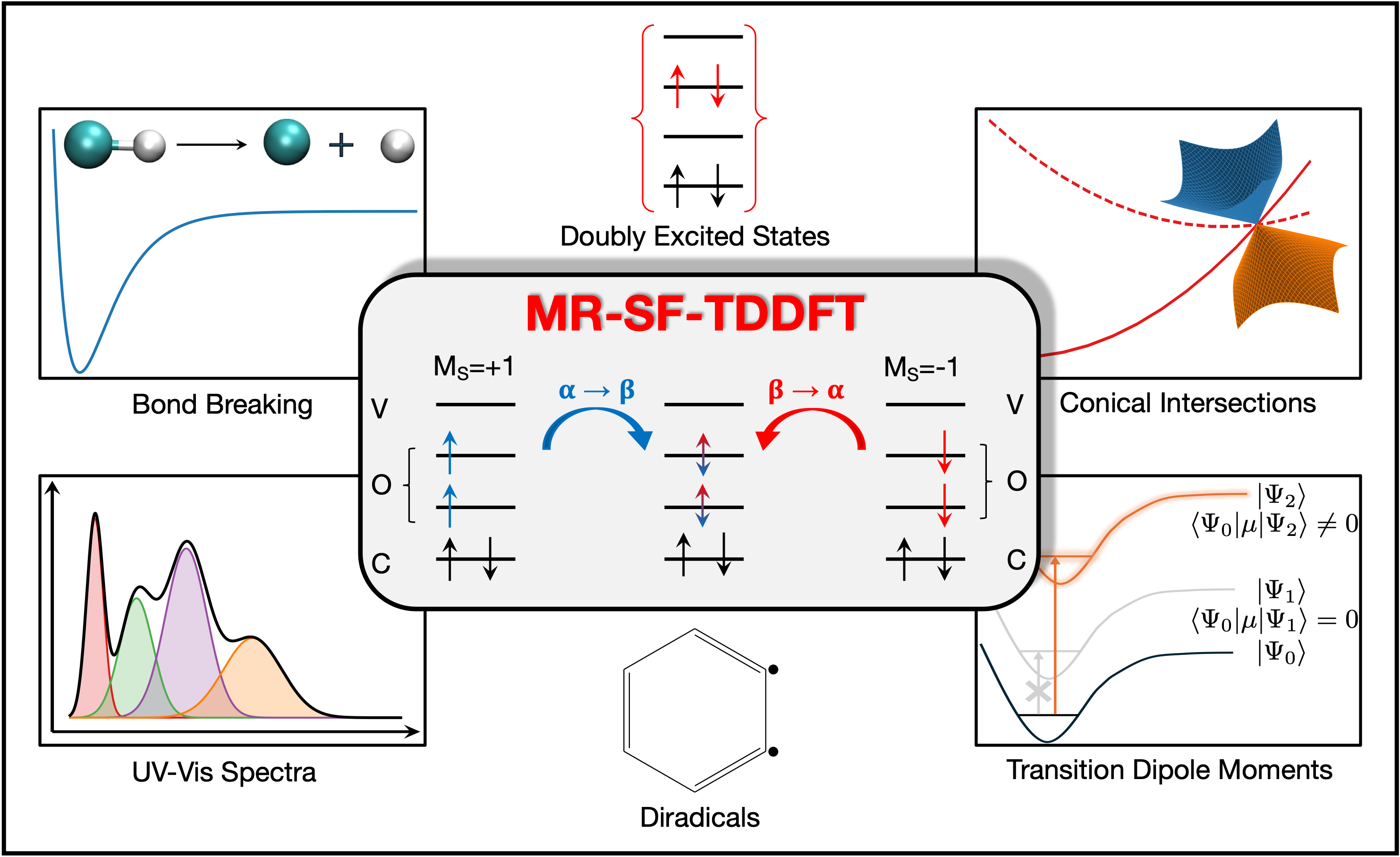
MRSF-TDDFT
MRSF-TDDFT is an improved version of spin-flip TDDFT that enables effectively spin-pure treatments of doubly excited states, bond-breaking, conical intersections, and some other cases of strongly correlated systems. Q-Chem 7 presents effective MRSF-TDDFT implementation including calculation of state and transition properties, such as state and transition dipole moments, oscillator strengths, spin–orbit couplings, and density-matrix based analyses of the MRSF states and transitions. Read the paper here.
New Analysis Tools
A new charge-transfer metric for TDDFT in libwfa. These metrics are rigorously invariant with respect to orbital rotations, unlike earlier metrics such as Tozer’s Lambda, and metrics widely used in the Gaussian program.
Energy decomposition analysis (EDA) can be used to decompose the energy into physically-meaningful components, and now includes ROKS-EDA, EDA OVOCV analysis, and force decomposition analysis (FDA).
New Spectroscopy Tools
- ECD for TDDFT and EOM-CCSD
- Restricted and unrestricted VCD
- CVS-XCIS for X-ray spectroscopy modeling
- New methods for modeling Auger decay
- New ΔSCF driver with easy-to-use input format and useful tools for analysis and visualization
QC-PBC: GTO-Based Solid-State Modeling
QC-PBC is a new module for handling solid-state systems and materials by all-electron calculations with periodic boundary conditions and Gaussian basis sets.
- DFT and TDDFT
- MP2, LT-MP2, and MP3 for gamma and k-point calculations
- CCSD, CCSD(T), and CCSDT for gamma-point calculations
- Python interfacing
- Geometry optimization
- Solvation
- Analytic frequency and phonon calculations
M-Chem: For Modeling Biological Systems
M-Chem is a new module for handling large biomolecular systems; it is slated for release in late July.
- High-performance MPI/OpenMP hybrid implementation for molecular dynamics (MD) simulations
- Fixed charge (Amber), polarizable (AMOEBA), and ReaxFF force fields
- Nose-Hoover thermostat and barostats, conjugate gradient self-consistent field and novel extended Lagrangian schemes for solving the many-body forces, periodic boundary conditions, and particle mesh Ewald
- Modular front-end that stream-lines parameter assignment and system preparation in a more user-friendly and reproducible way for standard force fields and protein-water simulations
- QM/MM integration with ReaxFF
- Trajectory formats for analysis in other codes
A One-Stop Shop for Computational Chemistry
Q-Chem 7.0 includes modules for molecular, material, and biomolecular modeling with no additional licensing.

Q-Chem Core Features
Density Functional Theory
- Vast built-in library of density functionals
- New local correlation methods for the fastest-ever double-hybrid DFT
- Great parallel performance
- New “Robust SCF” algorithm for easily converging tricky cases
- Solvation and Embedding
- Implicit solvent models, including SMD, C-PCM, and COSMO
- Explicit solvent modeling
- Density embedding methods
Excited States & Properties
- CIS
- TDDFT
- EOM-CC and ADC
Post-HF Methods
- Coupled-cluster and algebraic diagrammatic approaches
- RI-CC2
- Local correlation for MP2
- CASSCF, selected CI, and RAS-CI
- Spin-flip (including MRSF-TDDFT)
- Variational 2-RDM
Spectroscopy Modeling
- IR and UV-Vis spectroscopy
- Vibronic spectroscopy
- Photoelectron spectroscopy
- VCD and ECD spectroscopy
- NMR spectroscopy
- Non-linear spectroscopy
- Tools for X-ray spectroscopy: XPS, XAS, XES, Auger, RIXS, and more
Ready-To-Launch Cloud Capabilities
Q-Cloud provides fast and easy cloud computing through Amazon EC2, providing flexibility, scalability, and efficient performance for effective workflows.

Q-Chem Brochure

Ways To Try Q-Chem
IQmol is Q-Chem's open-source molecular editor and visualization package. Run Q-Chem jobs for free on our IQmol server!
Use Q-Cloud to easily run calculations in the cloud with AWS.
Request a free demo license to try Q-Chem for one month on your own hardware!