QC-PBC: Extending Q-Chem to Materials
Q-Chem is pleased to introduce QC-PBC: An effective and parallel GTO-based periodic code for accurate ab initio materials modeling. Core capabilities of QC-PBC include:
- Energies
- Band structures
- Geometry optimization
- Implicit solvation methods
- Fully analytic frequency and phonon calculations
- Energy decomposition analysis tools

Features Included in QC-PBC
Density Functional Theory
- Broad library of DFT functionals, including hybrid functionals
- OpenMP and MPI parallel implementations
- Fastest integral libraries
- GPW
- Density fitting
Post-HF Methods
- MP2, LT-MP2, and MP3
- BW-s2
- dRPA
- CCSD, CCSD(T), and CCSDT (gamma-point only)
Excited-State Methods
- CIS
- TDDFT
Analytic Frequency & Phonon Calculations
Solvation Methods
- LPCM
- NLPCM
- CANDLE
- Finite-temperature grand-canonical DFT for solid-liquid interfaces
Other Features
- Energy decomposition analysis (EDA)
- Scalar and relativistic effects via X2c1e
- Mulliken population analysis
- Localization methods
- Python interfaces enable interfacing to workflows and machine learning packages like PyTorch
A One-Stop Shop for Computational Chemistry
Q-Chem 7.0 includes modules for molecular, material, and biomolecular modeling with no additional licensing.

Recent QC-PBC Publications
Regularized Perturbation Theory for Ab Initio Solids. Meng-Fu Chen, Jinghong Zhang, Hieu Q. Dinh, Adam Rettig, and Joonho Lee. J. Phys. Chem. Lett. 2025, 16, 44, 11373–11381
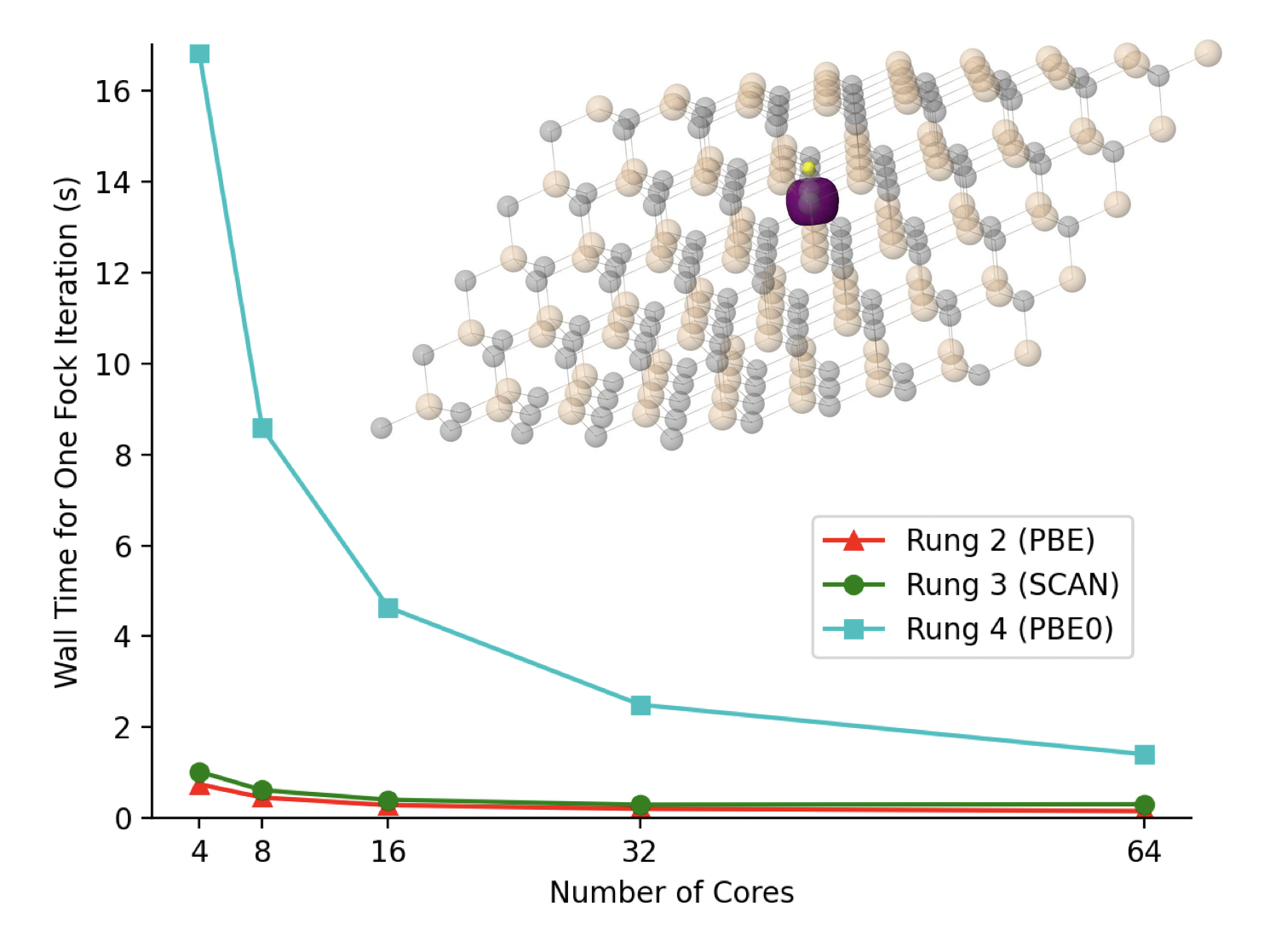
MP2 in QC-PBC: Authors developed regularized second-order perturbative methods in the QC-PBC package, including BW-s2, which provides accurate results for metals, semiconductors, and molecular crystals. Read the paper here.
Chemical Origins of Non-Bonded Interactions Within and Between Solids. Paul J. Robinson, Adam Rettig, Hieu Q. Dinh, Anton Z. Ni, and Joonho Lee. Preprint (arXiv). 2026.
ALMO-EDA in QC-PBC: In this recent preprint, the developers of QC-PBC extend ALMO-EDA to solid-state systems. They use this approach to study non-bonded interactions in solids, including molecular crystals, moiré heterobilayers, and layered perovskite heterostructures. They glean useful insights that allow them to approach materials design with clear, chemically-intuitive understanding.
Gaussian-Based Periodic Grand Canonical Density Functional Theory with Implicit Solvation for Computational Electrochemistry. Anton Z. Ni, Adam Rettig, and Joonho Lee. Preprint (arXiv). 2025.
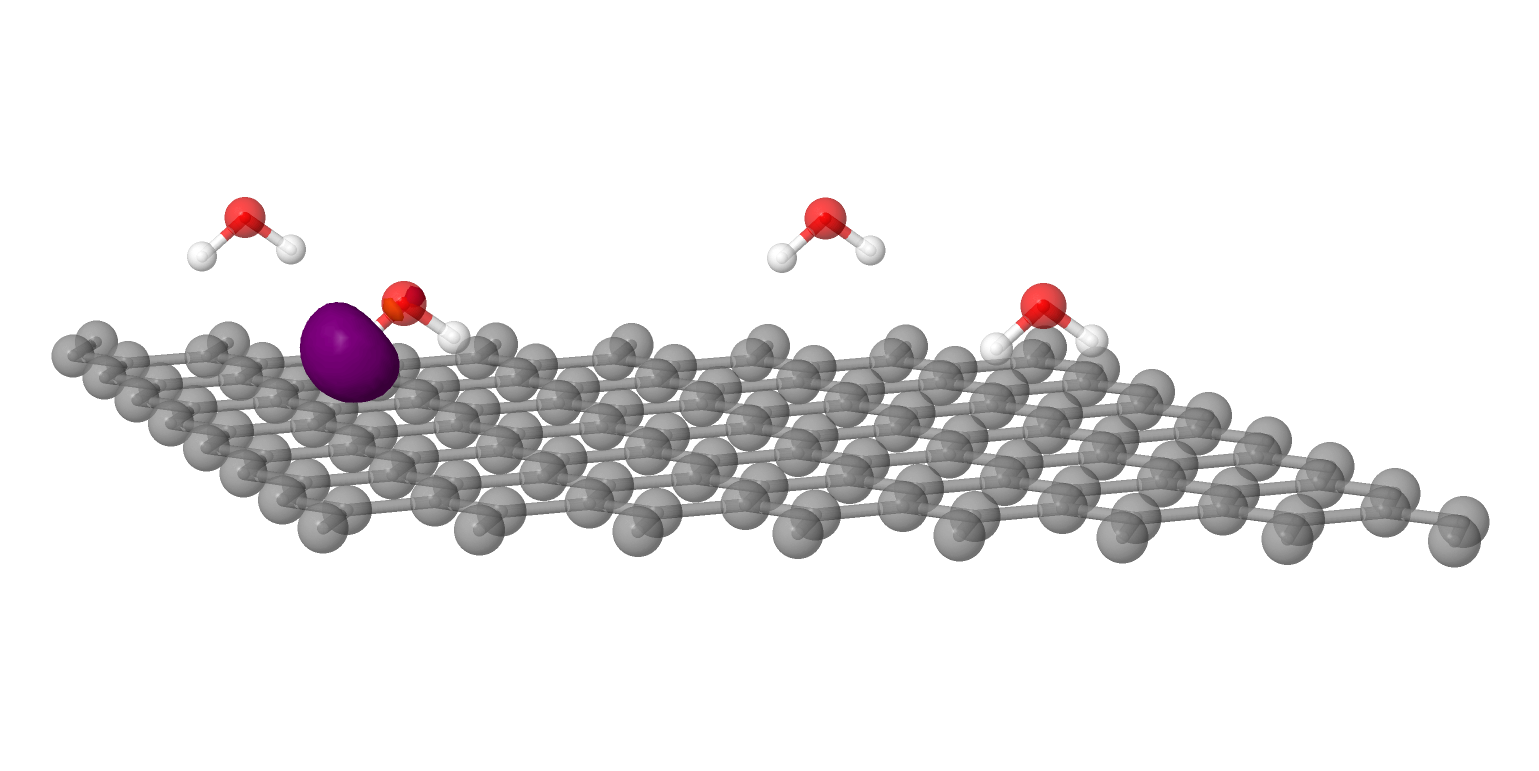
Modeling Electrochemistry at Solid-Liquid Interfaces: QC-PBC was recently used to model electrochemical reactions at solid-liquid interfaces! Developers introduced a numerical method for grand canonical density functional theory (DFT) for periodic systems in QC-PBC and then used their implementation, along with their implicit solvent modeling implementation, to model corrosion at silver surfaces.
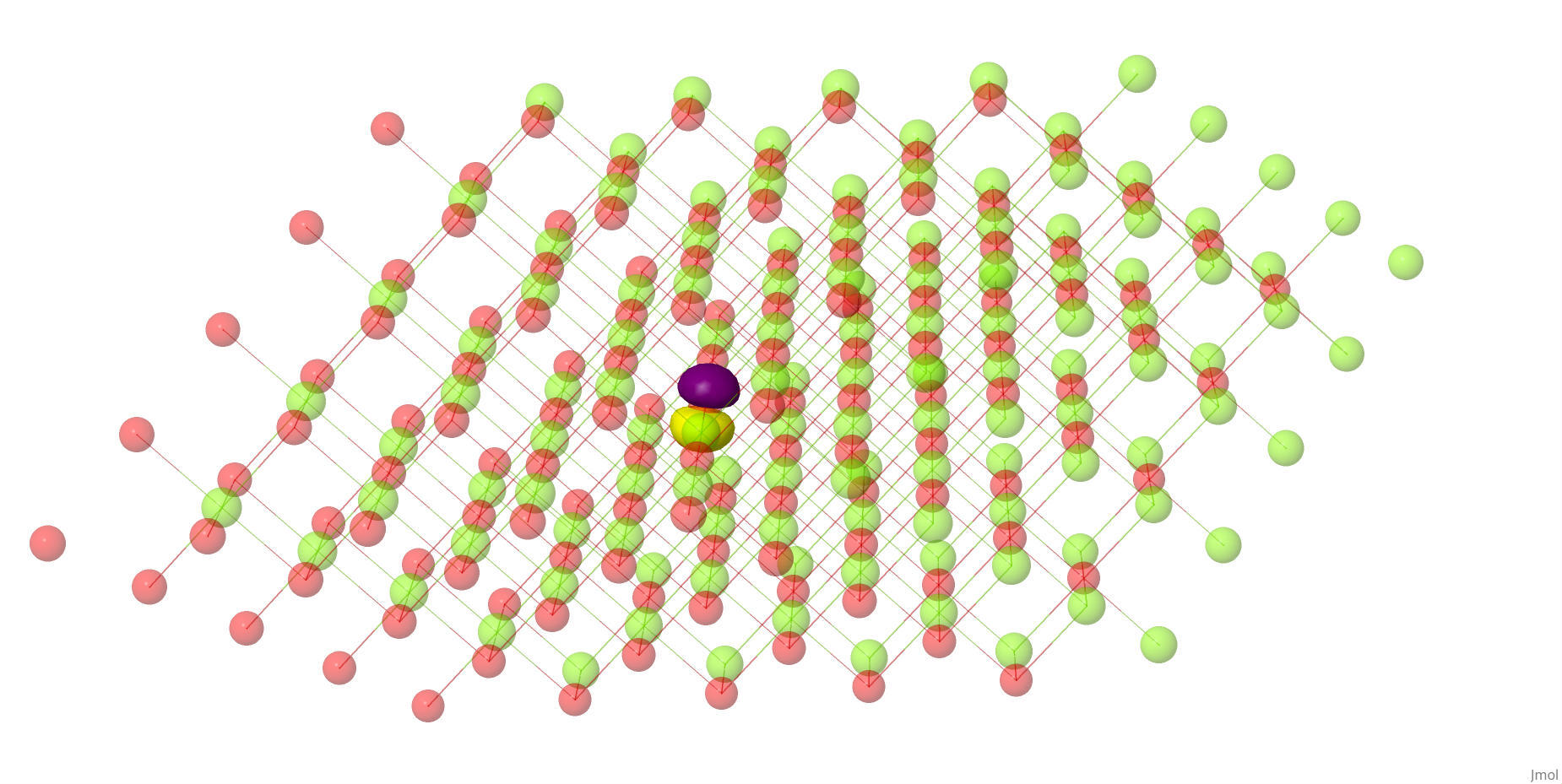
Intrinsic bond orbitals (IBOs) for MgO.

QC-PBC Webinar

Try QC-PBC for Free
Request a free demo license to try QC-PBC for one month on your own hardware!